2 min read
Connect with Veristat at Bio International Convention 2026
June 22-25, 2026 – San Diego, CA
🌎 Bringing the Global Biopharma Community Together
We’re delighted to be attending...

2 min read
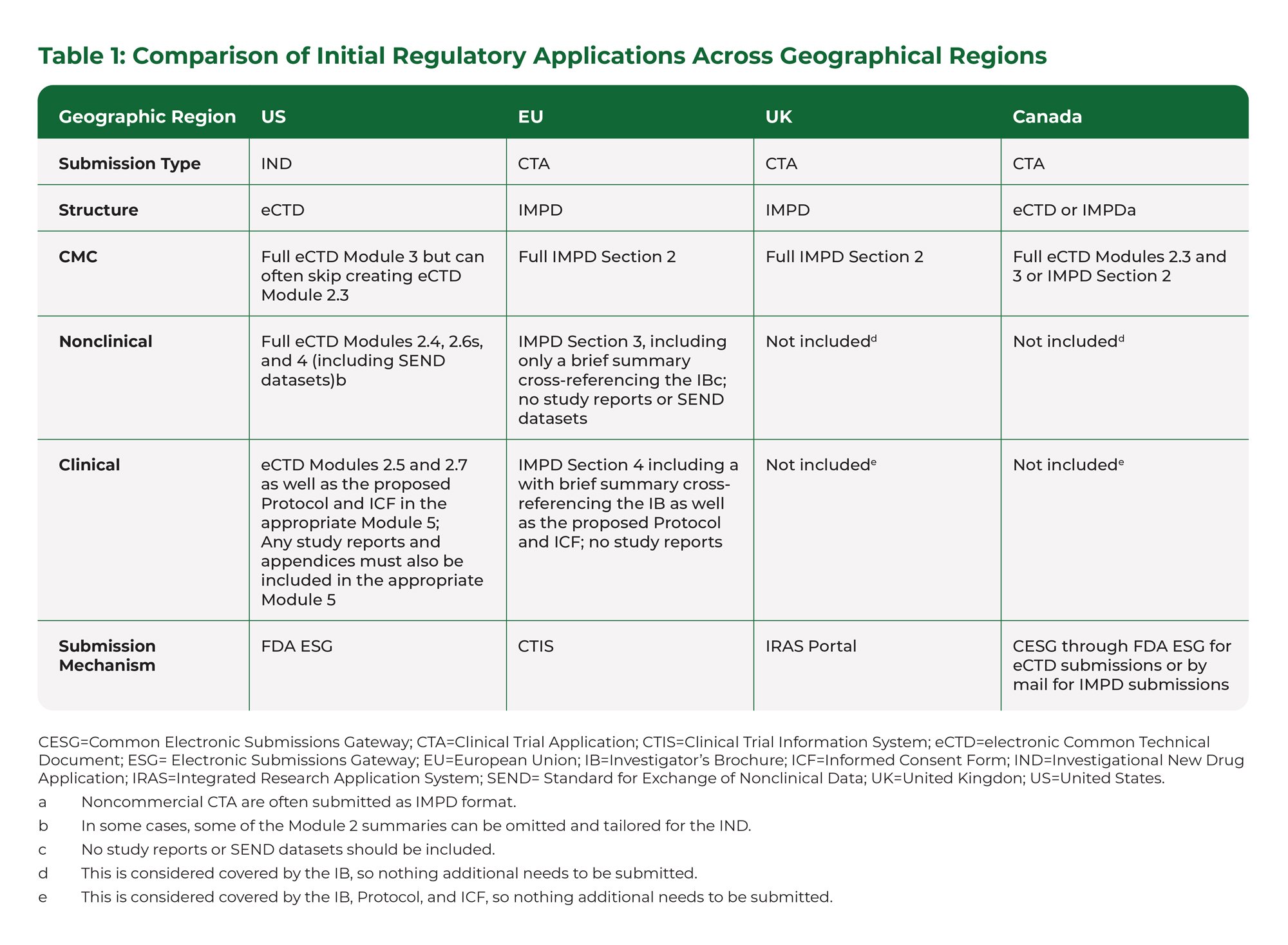
Investigational New Drug Applications (INDs) and Clinical Trial Applications (CTAs) are regulatory submissions needed for the initiation of clinical trials, and these applications have commonalities and differences throughout the world. If clinical trials are being initiated in multiple countries, then planning for these different submissions is critical to efficiency. In this paper, a comparison of the authoring of documents included in these submission types to the United States (US), European Union (EU), United Kingdom (UK), and Canada is discussed (Table 1 below), along with insights and recent regulatory updates.
An IND is used to initiate clinical trials in the US and is submitted to the Food and Drug Administration in electronic Common Technical Document (eCTD) format. There are limited Module 2 summaries depending on the product history. All underlying source reports, including Standard for Exchange of Nonclinical Data (SEND) datasets for applicable nonclinical studies, must be included. Luckily, once submitted for a specific drug and indication, all future studies, documents, and data for that drug and indication are submitted as additions to that same IND.
A CTA is used for initiation of clinical trials throughout most of the rest of the world. Most countries accepted chemistry, manufacturing, and controls (CMC), nonclinical, and clinical information summarized in an Investigational Medicinal Product Dossier (IMPD). As of 31 January 2023, the EU only accepts CTAs that are submitted via the Clinical Trial Information System (CTIS). A CTIS submission includes the IMPD as a component and cannot accommodate eCTD sections or structure. An added benefit of CTIS is that applications for up to 30 countries can be requested with the same documentation. Also, healthcare providers and patients can search public portions of the CTIS database for information.
There was no change to CTA submission requirements outside of the EU (i.e., there was no change for either the UK or Canada). The CMC section needs to be complete (equivalent quantity and level of information as in Module 3 in eCTD), and source reports are not necessary. The nonclinical and clinical sections can be very brief, high level summaries that largely cross-reference the Investigator Brochure (IB). All of this information is submitted each time the Sponsor wishes to initiate a new clinical trial, regardless of whether a CTA has already been submitted for a specific drug and indication. However, subsequent studies can cross-refer to the initial CTA, especially for the same CMC material.
For all 4 geographic areas discussed in this paper, the IB, Protocol, and Informed Consent Form are submitted in full in each region.
It is important to carefully plan for submission to multiple countries. The decision on which country to submit to first will have a waterfall effect on subsequent decisions such as eCTD, IMPD, or both formats (if submitting an IND and CTA) as well as how to most efficiently develop the CMC, nonclinical, and clinical content. For example, if a CTA will be submitted first, then the nonclinical information will be within the IB, while an IND will require completion of the eCTD Module 2 summaries. Conversion from a CTA to an IND is more complex and requires more time and effort than the reverse.
Veristat can assist you with your submission needs and can tailor your application to your unique clinical development program.